
By MacKenzie Griffith and Peter Lio, MD
Introduction
Epidermolysis bullosa (EB) describes a group of genetic diseases characterized by extremely fragile skin that forms blisters, erosions, and scars when exposed to minimal mechanical trauma.1 This condition can severely impact the quality of life for patients and caregivers. In addition to a reduced lifespan, patients with EB can have anxiety, depression, chronic pain, chronic itch, and a reduced capability to perform many activities of daily living.2 Caregivers who provide wound care face significant strain due to the time required to properly care for chronic wounds, the cost of wound care supplies, and the psychosocial impact of putting aside their personal needs for patients.3
A prospective cross-sectional study investigated the epidemiology of EB in the United States using data from more than 3,000 patients with EB contained within the National Institutes of Health National Epidermolysis Bullosa Registry (NEBR) between 1986 to 2002.4 In 2002, the prevalence of EB was 11.1 per 1 million live births. The incidence between 1986 and 2002 was 19.57 per 1 million live births.4 In this brief review, we discuss the most up-to-date thinking about the disease with regard to diagnosis, genetics, testing, and treatment.
Diagnosis
There is a wide range of clinical severity within EB, which can be largely attributed to the genotypic heterogeneity associated with the condition.5 Patients may exhibit a mild phenotype with a few blisters appearing on the hands and feet as a newborn, with few to no symptoms as an adult. Conversely, patients with severe forms of EB may experience widespread blistering that may result in physical disability. The skin changes can also impact the hair and nails, resulting in hair loss, nail fragility, or nail thickening. Some forms of EB may be accompanied by extracutaneous symptoms such as reno-urinary tract anomalies, muscular dystrophy, or pyloric atresia.6
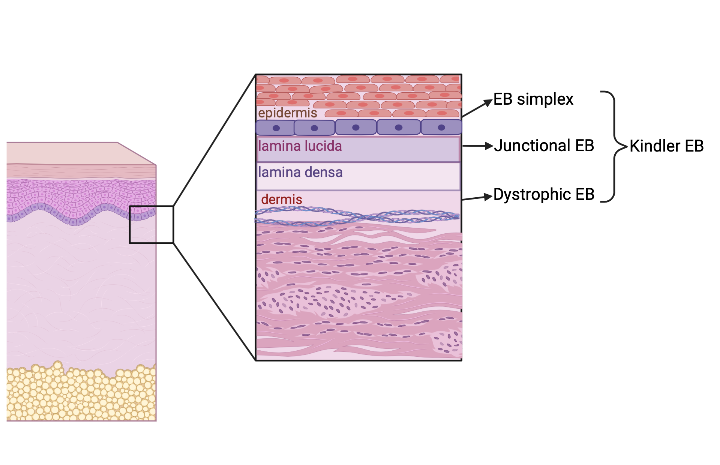
Because of this clinical variation, EB can be difficult to diagnose. An “onion skin” approach to diagnosis was proposed by Fine et al in 2014 .6,7 This approach begins with categorizing the EB into one of four clinical subtypes (Figure 1). The EB clinical subtype is based on the layer of skin where the blistering occurs, which can be visualized with a skin biopsy.1 EB simplex demonstrates cleavage between basal keratinocytes, leading to more superficial blisters and erosions (Figure 2). Junctional EB occurs when there are breaks within the lamina lucida of the basement membrane that separates the epidermis from the dermis.6 Dystrophic EB demonstrates cleavage beneath the basement membrane.6 Kindler EB has a mixed skin cleavage pattern.6
The next step to diagnosis in the aforementioned “onion skin” approach is evaluating the clinical phenotype, notably the distribution and severity of blistering.7
Figure 1: “Onion skin” diagnosis as proposed by Fine et al. (Figure made in biorender.com)
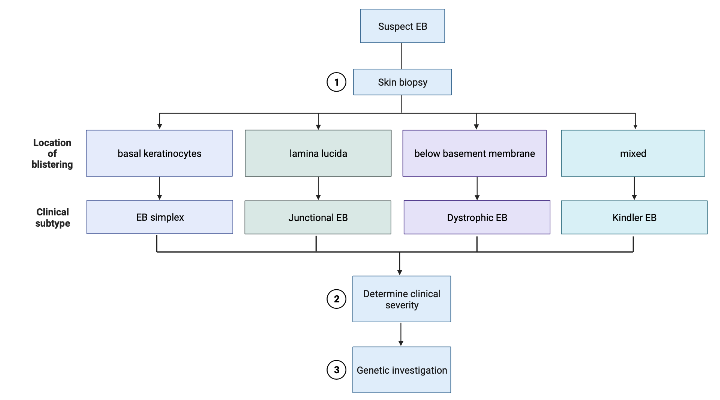
Figure 2: EB clinical subtypes (Figure made in biorender.com)
Genetics
The final step in EB diagnosis is genetic investigation. EB arises from mutations in structural components in various layers of the skin.8 Blister location within the skin is correlated with the mutation causing the structural defect. Possible components affected by these mutations include keratins, laminin, and type VII and XVII collagen. EB subtypes may demonstrate allelic heterogeneity, with similar phenotypes arising from mutations in different genes (Table 1). In EB simplex, blisters often arise from mutations in desmosomes, hemidesmosomes, or keratinocytes. Junctional EB is caused by mutations in hemidesmosomes. Hypoplastic anchoring fibrils cause dystrophic EB. Kindler EB is caused by mutations in the keratinocyte protein kindlin, also known as fermitin.
Table 1: Genes associated with EB clinical subtypes
| Affected genes based on clinical subtype5 | |||
| EB simplex | Junctional EB | Dystrophic EB | Kindler EB |
| Keratin 5, 14 | Laminin 332 | Type VII collagen | Kindlin-1 |
| Plectin | Type XVII collagen | Lysyl hydroxylase 3 | |
| Kelch-like protein 24 | Laminin α3A | ||
| BPAG1 | Integrin α6β4 | ||
| Exophilin 5 | Integrin α3 subunit | ||
| Tetraspanin 24 | |||
| Transglutaminase 5 | |||
| Plakophilin 1 | |||
| Desmoplakin | |||
| Plakoglobin | |||
Testing
Diagnosis of EB is confirmed by immunofluorescence (IF) mapping, transmission electron microscopy, or direct genetic analysis.5,6 Testing begins with obtaining a skin biopsy from the patient. The tissue is then exposed to monoclonal antibodies that target proteins in the dermo-epidermal junction that can be impacted by mutated proteins. If these antibodies bind to a mutated protein, they will demonstrate immunofluorescence and are visualized under a microscope.9 The presence or absence of IF can clue pathologists into the specific genetic cause of a patient’s EB. This testing method is often used to diagnose patients with greater disease severity since patients with mild EB may not demonstrate cleavage in a skin biopsy. Clinicians may start with IF mapping and then pursue genetic analysis to ensure accurate subclassification.
Transmission electron microscopy (TEM) uses skin biopsy samples to identify and quantify ultrastructural elements in the skin such as anchoring filaments, desmosomes, hemidesmosomes, and keratin filaments.1,9IF mapping is used more often and is often more accurate in diagnosing the subtype of EB than TEM.9
Direct genetic analysis looks at a patient’s genome to identify specific known mutations associated with EB. Multiple genes can be sequenced to look for a genetic cause, and clinicians can add less common genes to the panel if they are clinically suspected.1,6 Genetic analysis can be more expensive and time-intensive than IF mapping and TEM especially if the gene is not common. In this case, the entire exome may need to be analyzed to find the underlying genetic mutation. Multiple gene panels can make this form of testing more cost-effective. If these disease-specific methods do not reveal a mutation, a larger deletion or duplication may be suspected. Such changes may be investigated via quantitative real-time PCR or multiplex ligation-dependent probe amplification (MLPA). RNA sequence analysis may be used to detect splice sites within introns and requires RNA isolate from skin samples or cultured keratinocytes. Direct genetic analysis will become more clinically important as molecular treatments for EB become available.
Therapy
Presently, there is no curative therapy available for EB, only symptomatic treatment. The foundation of therapy is centered on combined wound care, proper nutrition, and avoiding excess mechanical trauma and sun exposure. Immunosuppressive medications including corticosteroids, azathioprine, cyclosporine, cyclophosphamide, mycophenolate mofetil, and methotrexate have been effective in reducing inflammation associated with EB.10 A combination of dapsone and prednisolone has been used to treat EB in children.11Severe, refractory cases of EB recalcitrant to traditional immunosuppression has been treated with rituximab and intravenous immunoglobulins (IVIG).12 EB pruriginosa, a subtype of dystrophic EB featuring severe pruritus, lichen simplex chronicus-like lesions, and prurigo nodularis, has been successfully treated with the IL4α receptor antagonist dupilumab (Dupixent, Sanofi, and Regeneron).13 High doses of colchicine have also been shown to be effective in treating EB.14 Colchicine is thought to reduce the production of antibodies and prevent antigen presentation to T cells, thereby reducing the immune response in EB.14
Newer approaches are under investigation for the treatment of EB. Current strategies being studied include gene therapy, cell therapy, RNA targeting therapy, and protein therapy. Benefits of gene therapies such as gene replacement or editing include long-term correction of disease symptoms and personalized therapy. Gene therapy is highly complex, can cause tumorigenesis, and can trigger an immunological response.14 Cell therapy such as revertant cell therapy, bone marrow stem cell transplant, and fibroblast stem cells also offer long-term disease correction and custom, personalized therapy and these generally carry a smaller risk of immunological response. RNA and protein therapy do not offer long-term symptom relief but are considered less invasive than gene and cell therapy.15
Conclusion
While EB remains a complex group of conditions with a multitude of known genetic causes and phenotypes, new diagnostic approaches provide a framework to help with this previously difficult-to-diagnose condition. Despite many known causes of EB, new genetic causes and phenotypes continue to be uncovered. There are a multitude of tests to diagnose EB and some may be tailored based on clinical suspicion for a specific subtype. Although there are not currently any curative therapies available for EB, exciting treatments are being tested that have the potential to dramatically improve patients’ quality of life, mental health, and lifespan. Fortunately, with proper funding and support, the future of EB diagnosis and treatment looks bright.
About the authors
C o
o
MacKenzie Griffith is a fourth-year medical student at Rush Medical College in Chicago, IL

Peter Lio, MD
Peter A. Lio, MD is a Clinical Assistant Professor of Dermatology and Pediatrics at Northwestern University Feinberg School of Medicine and a partner at Medical Dermatology Associates of Chicago.
Disclosures: None
References
- Mariath LM, Santin JT, Schuler-Faccini L, Kiszewski AE. Inherited epidermolysis bullosa: update on the clinical and genetic aspects. An Bras Dermatol. 2020;95(5):551-569. doi:10.1016/j.abd.2020.05.001
- Bruckner AL, Losow M, Wisk J, et al. The challenges of living with and managing epidermolysis bullosa: insights from patients and caregivers. Orphanet J Rare Dis. 2020;15(1):1. Published 2020 Jan 3. doi:10.1186/s13023-019-1279-y
- Chateau AV, Blackbeard D, Aldous C. The impact of epidermolysis bullosa on the family and healthcare practitioners: a scoping review. Int J Dermatol. 2023;62(4):459-475. doi:10.1111/ijd.16197
- Fine JD. Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates From the National Epidermolysis Bullosa Registry. JAMA Dermatol. 2016;152(11):1231-1238. doi:10.1001/jamadermatol.2016.2473
- Has C, Liu L, Bolling MC, et al. Clinical practice guidelines for laboratory diagnosis of epidermolysis bullosa. Br J Dermatol. 2020;182(3):574-592. doi:10.1111/bjd.18128
- Has C, Fischer J. Inherited epidermolysis bullosa: New diagnostics and new clinical phenotypes. Exp Dermatol. 2019;28(10):1146-1152. doi:10.1111/exd.13668
- Fine JD, Bruckner-Tuderman L, Eady RA, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70(6):1103-1126. doi:10.1016/j.jaad.2014.01.903
- Has C, Bruckner-Tuderman L. The genetics of skin fragility. Annu Rev Genomics Hum Genet. 2014;15:245-268. doi:10.1146/annurev-genom-090413-025540
- Yiasemides E, Walton J, Marr P, Villanueva EV, Murrell DF. A comparative study between transmission electron microscopy and immunofluorescence mapping in the diagnosis of epidermolysis bullosa. Am J Dermatopathol. 2006;28(5):387-394. doi:10.1097/01.dad.0000211510.44865.6d
- Hallel-Halevy D, Nadelman C, Chen M, Woodley DT. Epidermolysis bullosa acquisita: update and review. Clin Dermatol. 2001;19(6):712-718. doi:10.1016/s0738-081x(00)00186-3
- Kim JH, Kim YH, Kim SC. Epidermolysis bullosa acquisita: a retrospective clinical analysis of 30 cases. Acta Derm Venereol. 2011;91(3):307-312. doi:10.2340/00015555-1065
- Crichlow SM, Mortimer NJ, Harman KE. A successful therapeutic trial of rituximab in the treatment of a patient with recalcitrant, high-titre epidermolysis bullosa acquisita. Br J Dermatol. 2007;156(1):194-196. doi:10.1111/j.1365-2133.2006.07596.x
- Shehadeh W, Sarig O, Bar J, Sprecher E, Samuelov L. Treatment of epidermolysis bullosa pruriginosa-associated pruritus with dupilumab. Br J Dermatol. 2020;182(6):1495-1497. doi:10.1111/bjd.18855
- Cunningham BB, Kirchmann TT, Woodley D. Colchicine for epidermolysis bullosa acquisita. J Am Acad Dermatol. 1996;34(5 Pt 1):781-784. doi:10.1016/s0190-9622(96)90013-4
- Prodinger C, Reichelt J, Bauer JW, Laimer M. Epidermolysis bullosa: Advances in research and treatment. Exp Dermatol. 2019;28(10):1176-1189. doi:10.1111/exd.13979
