Soligenix, Inc. has gotten a green light from the European Medicines Agency (EMA) on the key design components of a confirmatory Phase 3 placebo-controlled study evaluating the safety and efficacy of synthetic hypericin (HyBryte) for the treatment of cutaneous T-cell lymphoma (CTCL) patients with early-stage disease.
HyBryte (SGX301) is a novel, first-in-class, photodynamic therapy utilizing safe, visible light for activation. The active ingredient in HyBryte is synthetic hypericin, a potent photosensitizer that is topically applied to skin lesions taken up by the malignant T-cells, and then activated by safe, visible light approximately 24 hours later. HyBryte has received orphan drug and fast-track designations from the U.S. Food and Drug Administration (FDA and orphan designation from the EMA.
This confirmatory 18-week study is expected to enroll approximately 80 patients in the United States and Europe and is targeted to begin patient enrollment by the end of 2024 with top-line results anticipated in the second half of 2026. Discussions with the FDA on an appropriate study design remain ongoing. The agency has expressed a preference for a longer-duration comparative study over a placebo-controlled trial.
The new EMA-approved study, FLASH2 (Fluorescent Light Activated Synthetic Hypericin 2), replicates the double-blind, placebo-controlled design used in the first successful Phase 3 FLASH study that consisted of three six-week treatment cycles (18 weeks total), with the primary efficacy assessment occurring at the end of the initial six-week double-blind, placebo-controlled treatment cycle (Cycle 1). However, the second study extends the double-blind, placebo-controlled assessment to 18 weeks of continuous treatment (no “between-cycle” treatment breaks) with the primary endpoint assessment occurring at the end of the 18-week timepoint. In the first Phase 3 study, a treatment response of 49% was observed in patients completing 18 weeks (three cycles) of therapy. In this second study, all important clinical study design components remain the same as in the first FLASH study, including the primary endpoint and key inclusion-exclusion criteria. The extended treatment for a continuous 18 weeks in a single cycle is expected to statistically demonstrate HyBryte’s increased effect over a more prolonged, “real world” treatment course.
“In treating CTCL, which is a chronic cancer with no cure, long-term safety is a strong driver of treatment choice. Most current treatment options for CTCL are associated with significant safety concerns, including black-box warnings,” says Brian Poligone, MD, PhD, Director of the Rochester Skin Lymphoma Medical Group in Fairport, NY, in a news release. “Clinical studies with HyBryte have demonstrated strong and rapid efficacy with a very benign safety profile, with broad applicability across different lesion types, different skin tones, and different disease stages. I know I can speak for my colleagues who have been involved with these studies when I say that the data generated to date has been extremely compelling. This second study is very similar to the first FLASH study and should build on these compelling data while allowing us to more closely as we would in a ‘real world’ setting. We believe the outcome of this trial will further validate the utility of HyBryte in early-stage CTCL and we look forward to participating in this important study.”
“With its chronic course and major impact on patient quality of life, CTCL is an orphan disease in urgent need of additional treatment options that are well-tolerated and safe over the long term,” adds Christopher Schaber, PhD, President and Chief Executive Officer of Soligenix. “Studies to date have indicated a substantial increase in efficacy with longer treatment and similar performance against both patch and plaque lesions. These results are derived from one of the largest studies ever conducted in CTCL, and we believe this second study will both substantiate and improve upon these results.”
In the confirmatory Phase 3 study, HyBryte will be topically applied to CTCL lesions twice weekly for 18 weeks, with each application followed 21 (±3) hours later by the administration of safe, visible light at a wavelength of 500 to 650 nm. The light will be administered starting at 6 J/cm2 twice weekly. This will be increased upwards by 2 J/cm2 until 1) the patient experiences a Grade 1 erythema, 2) the patient reaches the maximum dose of 30 J/cm2, or 3) the patient cannot tolerate the treatment time, whichever comes first.
All of the patient’s lesions that are readily available for exposure to the visible light source will be treated and 3 to 5 index lesions in each patient will be prospectively identified and indexed for the modified composite assessment of index lesions severity (mCAILS) evaluation before randomization (baseline). The primary efficacy endpoint will be assessed on the percent of patients in each of the two treatment groups (HyBryte and placebo) achieving a Partial or Complete Response (yes/no) of the treated lesions defined as a ≥ 50% reduction in the total mCAILS score for the 3-5 index lesions following 18 weeks of treatment compared to the total mCAILS score at baseline. Other secondary measures will assess treatment response (including duration), degree of improvement, time to relapse, and safety. Following treatment, all patients will be followed every 4 weeks for a total of 12 weeks (through Week 30). The Data Monitoring Committee (DMC) will conduct one interim analysis when approximately 60% of the total subjects have completed the primary endpoint evaluation. The primary efficacy endpoint and the key safety endpoints will be analyzed. A sample size recalculation may be performed after examining the assumptions or the trial halted for either futility, safety concerns, or overwhelming efficacy. Soligenix, participating clinical investigators, and any personnel involved in trial conduct will remain blinded until the completion of the trial.
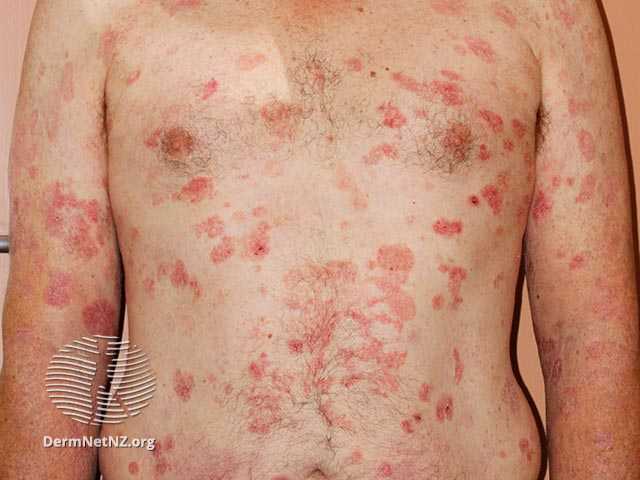
The published Phase 3 FLASH trial enrolled a total of 169 patients (166 evaluable) with Stage IA, IB, or IIA CTCL. The trial consisted of three treatment cycles. Treatments were administered twice weekly for the first six weeks and treatment response was determined at the end of the 8th week of each cycle. In the first double-blind treatment cycle (Cycle 1), 116 patients received HyBryte treatment (0.25% synthetic hypericin), and 50 received placebo treatment of their index lesions. A total of 16% of the patients receiving HyBryte achieved at least a 50% reduction in their lesions (graded using a standard measurement of dermatologic lesions, the CAILS score) compared to only 4% of patients in the placebo group at eight weeks during the first treatment cycle (primary endpoint). HyBryte treatment in this cycle was safe and well tolerated.
In the second open-label treatment cycle (Cycle 2), all patients received HyBryte treatment of their index lesions. Evaluation of 155 patients in this cycle (110 receiving 12 weeks of HyBryte treatment and 45 receiving six weeks of placebo treatment followed by six weeks of HyBryte treatment), demonstrated that the response rate among the 12-week treatment group was 40%. Comparison of the 12-week and six-week treatment groups also revealed a statistically significant improvement between the two groups, indicating that continued treatment results in better outcomes. HyBryte continued to be safe and well-tolerated. Additional analyses also indicated that HyBryte is equally effective in treating both plaque and patch lesions of CTCL, a particularly relevant finding given the historical difficulty in treating plaque lesions in particular.
The third (optional) treatment cycle (Cycle 3) was focused on safety and all patients could elect to receive HyBryte treatment for all their lesions. Of note, 66% of patients elected to continue with this optional compassionate use/safety cycle of the study. Of the subset of patients that received HyBryte throughout all 3 cycles of treatment, 49% of them demonstrated a positive treatment response. Moreover, in a subset of patients evaluated in this cycle, it was demonstrated that HyBryte is not systemically available, consistent with the general safety of this topical product observed to date. At the end of Cycle 3, HyBryte continued to be well tolerated despite extended and increased use of the product to treat multiple lesions.
The overall safety of HyBryte is a critical attribute of this treatment and was monitored throughout the three treatment cycles (Cycles 1, 2, and 3) and the 6-month follow-up period.
PHOTO CREDIT: DermNetNZ
