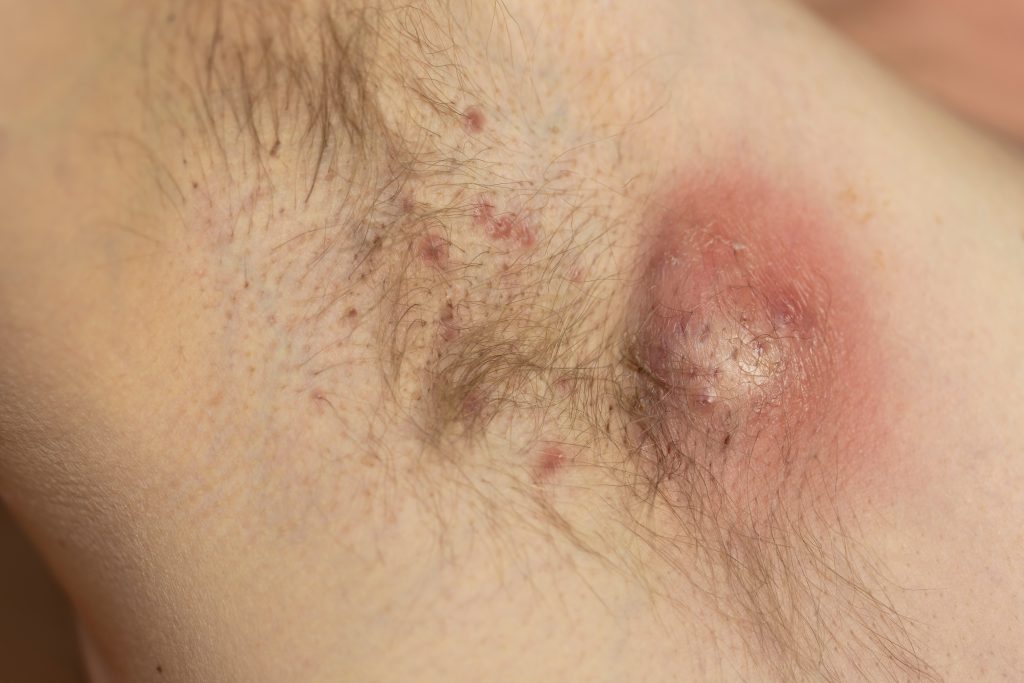
Povorcitinib (Incyte) produced statistically significant results for primary endpoints in two Phase 3 Hidradenitis Suppurativa (HS) trials.
Povorcitinib (INCB54707) is an oral small-molecule Janus kinase (JAK) 1 inhibitor currently in Phase 3 clinical trials for HS, vitiligo, and prurigo nodularis (PN), as well as Phase 2 trials for asthma and chronic spontaneous urticaria (CSU). The new data will support planned regulatory submission for povorcitinib in HS worldwide.
Both Met Primary Endpoint
Both the STOP-HS1 and STOP-HS2 studies met their primary endpoint at both tested doses (45mg and 75mg). A significantly higher proportion of patients treated with povorcitinib once daily (QD) vs. placebo achieved Hidradenitis Suppurativa Clinical Response (HiSCR), a ≥50% reduction from baseline in the total abscess and inflammatory nodule count (AN count), with no increase from baseline in abscess or draining tunnel count. The percentage of povorcitinib-treated patients achieving HiSCR50 compared to placebo at Week 12 was:
| STOP-HS1: | 45mg: 40.2% vs. 29.7%
75mg: 40.6% vs 29.7% |
| STOP-HS2:
|
45mg: 42.3% vs. 28.6%
75mg: 42.3% vs. 28.6% |
Within a predefined subgroup of patients previously exposed to biologics, povorcitinib demonstrated greater differential efficacy (HiSCR50) when compared to placebo (nominal P-values):
| STOP-HS1: | 45mg: 34.2% vs. 21.9%
75mg: 37.8% vs. 21.9% |
| STOP-HS2:
|
45mg: 45.0% vs. 19.5%
75mg: 40.0% vs. 19.5% |
In addition, at Week 12, patients treated with povorcitinib achieved deep levels of clinical response with a greater proportion achieving HiSCR75, reduction in flares, >3-point decrease in the Skin Pain Numeric Rating Scale (NRS) score and Skin Pain NRS30. Furthermore, povorcitinib demonstrated rapid onset of response, including rapid skin pain reduction.
The overall safety profile of povorcitinib is consistent with previous data. No new safety signals were observed, and both doses were well-tolerated.
“Hidradenitis suppurativa is a challenging and debilitating condition without a cure. Given the limitations of current HS treatments and its impact on patients’ daily lives, there is a critical need for new, well-tolerated, and effective therapies that provide a rapid reduction in the signs and symptoms of HS, in particular, pain,” says Steven Stein, MD, Chief Medical Officer at Incyte, in a news release. “The positive Phase 3 data highlights the potential of povorcitinib as an effective oral treatment option for people living with HS.”
More About STOP-HS
The STOP-HS clinical trial program includes two Phase 3 studies, STOP-HS1 (NCT05620823) and STOP-HS2 (NCT05620836), evaluating the efficacy and safety of povorcitinib (INCB54707) in adult patients with moderate-to-severe HS. Both studies include a 12-week double-blind, placebo-controlled treatment period, followed by a 42-week extension period and a 30-day safety follow-up.
The studies have each enrolled approximately 600 patients (age ≥18 years) diagnosed with moderate-to-severe HS for at least three months prior to the screening visit and met certain criteria: total AN count of ≥5, lesions in at least two distinct anatomical areas, a documented history of inadequate response to at least a three-month course of at least one conventional systemic therapy (oral antibiotic or biologic drug) for HS, or demonstrated intolerance to, or a contraindication to, such conventional systemic therapies.
The primary endpoint for both studies is the proportion of patients who achieve HiSCR50, defined as at least a 50% reduction from baseline in the total AN count at Week 12, with no increase from baseline in abscess or draining tunnel count. Key secondary endpoints include the proportion of patients achieving a 75% reduction in AN count with no increase from baseline in abscess or draining tunnel count (HiSCR75) at Week 12, the proportion of patients experiencing at least one flare-up over 12 weeks, the proportion of patients with a >3-point decrease in the Skin Pain NRS score among those with a baseline score of ≥3, and the proportion of patients achieving a 30% reduction and at least 1-unit reduction from baseline in Skin Pain NRS at Week 12. The studies also evaluate the frequency and severity of adverse events during the study.
