Thomas Leung, MD, PhD, an Associate Professor at the University of Pennsylvania in Philadelphia, PA, is investigating the underlying mechanisms and immune responses involved in acute febrile neutrophilic dermatosis—better known as Sweet’s syndrome—to improve diagnosis and treatment strategies.
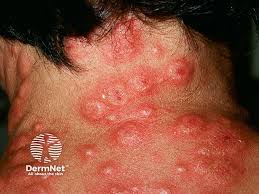
Sweet’s syndrome is characterized by fever, increased neutrophils in the blood, a painful skin rash, and, in some cases, multi-organ involvement with significant risk of increased mortality. “When you biopsy these lesions by [haematoxylin and eosin] H&E, you see a very characteristic neutrophilic infiltrate in the dermis,” Leung says during a plenary session at the annual meeting of the Society for Investigative Dermatology (SID) in San Diego, CA.
Researchers have proposed several possible pathways for the disease’s pathogenesis. “The first is a hypersensitivity reaction,” he says. “The skin is just irritated and causes more neutrophils to come in. Other people suggested a cytokine dysregulation, and there’s been some studies that suggested a genetic susceptibility as well.”
Report Shows Mutation
In 2023, Leung and colleagues published a report in the Journal of Clinical Investigation showing a 51-year-old woman with chronic Sweet’s syndrome that was resistant to treatment. They discovered that she had a mutation in her neutrophils in the PIK3R1 gene that allowed them to be more sensitively recruited to IL-1 compared to normal neutrophils.
“Because of this data, we gave her an IL-1 receptor inhibitor called Anakinra, which blocked this neutrophil migration and cleared her Sweet’s syndrome completely, which was great,” Leung says. “This might explain how neutrophils get into the skin, but neutrophils are thought to be very short-lived cells. They die within 48 to 72 hours, and we’re always puzzled by how they persist in the Sweet’s syndrome lesions themselves.”
To find out, Dr. Leung and two members of his lab, Satish Sati, PhD, and John Huang, PhD, collected skin and blood from six Sweet’s syndrome patients, four with classic or idiopathic Sweet’s syndrome and two with malignancy-associated Sweet’s syndrome, and performed single-cell RNA sequencing. They used four methods to confirm that they were honing on the correct subcluster of neutrophils. One was to gather all published single-cell datasets of bona fide neutrophils, some with cell sorting.
“We used a Spearman correlation plot to ask how close are our cells that we get in our single-cell cluster to these published single-cell clusters, [and found] almost 70% similarity among all these other datasets, making us quite confident that we have correctly identified neutrophils,” Dr. Leung recalls.
Neutrophils in Skin vs Blood
They discovered that neutrophils in the skin were activated differently from neutrophils in the blood and expressed antigen presentation (APC-like) genes. This activation state allowed skin neutrophils to live two to three times longer. Next, the researchers studied what signals were passed between keratinocytes and neutrophils in Sweet’s syndrome skin compared to unaffected skin.
“The highest signaling was a signaling pathway called SAA1 or serum amyloid A1 protein, signaling through the G protein-coupled receptor (GPCR) formyl peptide receptor 2 (FPR2),” he says. “When we looked at our single-cell dataset, SAA1 was only expressed in keratinocytes, while FPR2 was only expressed in neutrophils. This suggested that SAA1 and FPR2 signaling is what’s responsible for generating these long-lived neutrophils.”
What’s Driving the Disease
One question that keeps coming up, he adds, is: How do you think antigen-presenting cells (APC) neutrophils are really driving this disease? To date, Dr. Leung and his fellow researchers have observed that APC-like neutrophils are more effective at recruiting T cells compared with normal neutrophils.
“About five-fold more T cells are being recruited by APC-like neutrophils compared to normal neutrophils,” he says. “Also, these APC-like neutrophils are able to induce cytokine production from T cells, including IL-17.”
Based on their work so far, the researchers think that when neutrophils invade the skin, they irritate keratinocytes to begin to secrete SAA1. That SAA1 binds to a receptor on neutrophils called FPR2, which turns them into long-lived, APC-like cells that release more cytokines. This, in turn, draws in more T cells and prompts them to produce additional cytokines like IL-17.
“We think that by blocking SAA1-FPR2 signaling, this may represent the first targeted treatment for Sweet’s syndrome,” Dr. Leung concludes.
He disclosed that the research has been supported by the National Institutes of Health, the Department of Veterans Affairs, and the Berstein Foundation.
PHOTO CREDIT: DermNet
— Doug Brunk
